1.1.2. Draw Molecule¶
This page shows how to make molecular mapameter from the drawed molecule.
In the case that users want to make their drawed molecules, Users push the following image in PolyParGen front page.
Step1, users have to registrate their information to use PolyParGen. E-mail address certainly is needed in user’s informaiton, because PolyParGen sends that parameter file to user’s e-mail after generation of molecular parameter. The users, which already registrated, pass this step.
Step2, users draw a molecule under our drawer tool.

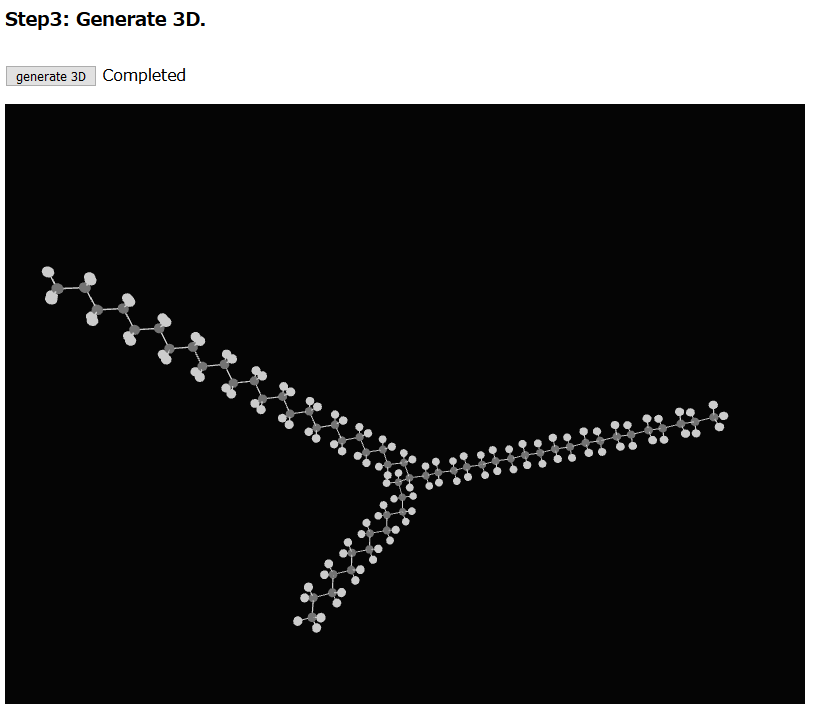
Step3, users confirm the drawn molecule in a 3D structure from with “generate 3D” button. If the molecule has a different structure, users draw the molecule again.
Step4, user setect the options for making molecular parameter.
This is the window of Step4.

Select PolyParGen version
At present, the support of PolyParGen version 1 has been suspended to create accurate potential parameters. Users select PolyParGen version 2 (v2).

Select the type of Lennard-Jones potential
Users select OPLS-AA or Amber parameter. In the case of Amber parameter, users select an atomic charge evaluation method (bcc or gas).

Set the number of fragment’s atoms
Users set the number of atoms contained in one fragment. The default value is 150 atoms. Basically, the default value is recommended.
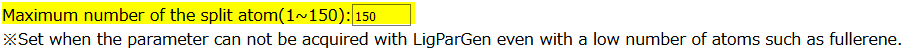
Extra options
For large-scale conjugated molecules such as graphene and fullerene, users have to set “0”.

Whether to evaluate atomic charges by ab initio calculation.
If users want to select the estimated atomic charge by ab initio calculation, users select this option. Also, users have to select the method, basis function and charge densith of method. ESP is the electrostatic potential fitting method. Mulliken is Mulliken density analysis.
Step5,Input the registrated e-mail address. After PolyParGen makes the moleculer parameter, the molecule’s parameter files are sent to the registrated e-mail address. Users have to input your e-mail.

Finaly, users push “Submit Molecule” button.